Time:2025-09-12
A new study published in Signal Transduction and Targeted Therapy on September 11, 2025, reports the discovery of SLC38A6 as a novel gene associated with essential tremor (ET). The research was a collaborative effort led by Prof. Jing-Yu Liu (Center for Excellence in Brain Science and Intelligence Technology, CAS), Prof. Beisha Tang (Xiangya Hospital, Central South University), Prof. Man Jiang (Tongji Medical College, HUST), and Prof. Jun Liu (Ruijin Hospital, Shanghai Jiao Tong University).
The team identified solute carrier family 38 member 6 (SLC38A6) as a novel ET-associated gene in a five-generation family from Tianjin with WES. Subsequent WGS in the ET cohort identified 15 distinct variants. Using both global and Purkinje cell-specific knockout mouse models (Slc38a6-/- and Slc38a6PC-/-), the researchers demonstrated that disruption of amino acid transporter encoded by SLC38A6 induces ferrpotosis in cerebellar Purkinje cell. These findings uncover a molecular and cellular mechanism contributing to ET and highlighting SLC38A6 as a promising therapeutic target for precision diagnosis and therapy.
Essential tremor (ET) is one of the most common neurological disorders, affecting about 1% of the general population, 5% of individuals older than 65 years, and over 20% of those older than 95 years. It is characterized by 4–12 Hz kinetic or postural tremors of the upper limbs, severely disrupting fine motor task such as writing, drinking and eating, abolishing the self-care ability. Because ET lacks reliable biomarkers and shows marked clinical heterogeneity, and diagnosis remains rely on clinical behavioral and exclusionary, which limits both accurate classification and research progress. Although many candidate genes and loci have been reported, the etiology of ET remains unclear.
To address these challenges, Prof. Jing-Yu Liu’s team at CEBSIT performed whole-exome sequencing in a five-generation ET pedigree and identified SLC38A6 as a novel candidate gene for ET (Fig. a and b). Whole-genome sequencing further uncovered 15 protein-altering variants in 772 familial ET probands and 640 sporadic individuals, including 71 (9.18%) Chinese families and 47 (7.34%) sporadic individuals with ET. These variants consisted of 13 missense and 2 splice-site mutations (Fig.c).
To validate function of SLC38A6 variants, the three most common human mutations (p.Y108F, p.M281T and p.G318S) were expressed in HeLa cells. Although the mutant SNAT6 transporters showed normal expression and localization, they significantly impaired L-arginine (L-Arg) uptake. In line with recent neuroimaging and postmortem data showing cerebellar pathology, including Purkinje cell, basket cell and climb fiber, the researchers generated global and Purkinje cell specific deletion mouse models (Slc38a6-/- and Slc38a6PC-/-). Both models showed ET-like tremor and Purkinje cell loss, dentritic and axonal degeneration, abnormal climber-Purkinje cell synapses, and basket cell axon overgrowth. Slice electrophysiology further demonstrated reduced neuronal Purkinje cell (PC) excitability and elevated inhibitory synaptic transmission, consistent with elevated “hairy” basket coverage around the PC soma. At the cellular level, PCs from knockout mice displayed mitochondrial abnormalities and elevated ferroptosis markers (ACSL4, TFRC and Fe ions). Finally, the researchers identified ATF3 upregulation as a possible driver of ferroptosis in Slc38a6-deficient PCs (Fig. d).
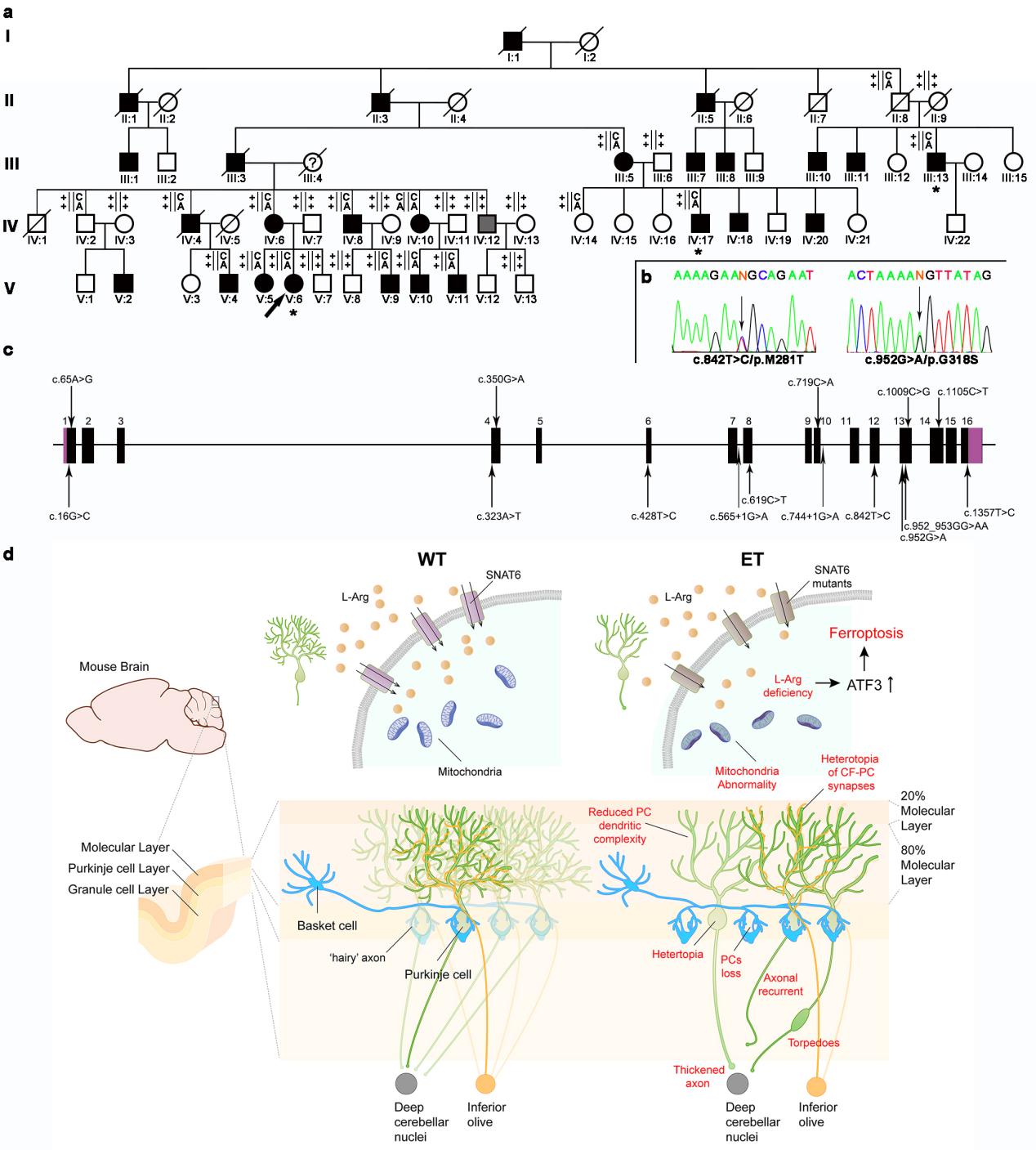
Figure legend: (a) Pedigree of family 1 and the genotypes of the family members. Squares and circles represent males and females, respectively. The filled and blank symbols indicate affected subjects with ET and asymptomatic subjects, respectively. The arrow denotes the proband of family 1. (b) Sanger sequencing results of the two variants of SLC38A6 identified in Family 1. Variant loci are indicated with arrows. (c) Schematic display of SLC38A6 with ET-associated variants indicated. (Upper)Schematic structure diagrams of SNAT6 with ET-associated mutations (blue circles) (down). (d) A working model for variants of SLC38A6 causing ET.
This study identified SLC38A6 variants in both familial and sporadic ET patients and uncovered how SNAT6 dysfunction contributes to cerebellar abnormalities. The Slc38a6 deletion mouse models mimic the tremors and pathological changes of ET, thereby revealing the molecular mechanisms driving the disease. This study provides novel molecular insights into ET pathogenesis and point to potential targets for future therapeutic strategies.
Contact:
LIU JIngyu
Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences, Shanghai, China.
liujy@ion.ac.cn
 附件下载:
附件下载: 
